算法:单细胞转录组测序联合bulk转录组测序_单核转录组测序
目录:
1.单细胞转录组测序技术原理及其应用
2.转录组测序和单细胞测序区别
3.转录组测序和单细胞转录组测序的区别
4.单个细胞的转录组测序
5.单细胞联合转录组找关键基因
6.单细胞转录组学技术
7.单细胞基因组与转录组
8.单细胞转录组测序的原理
9.单细胞转录组测序技术原理
10.单细胞转录组测序应用
1.单细胞转录组测序技术原理及其应用
近些年,单细胞技术的兴起为科研人员的精准研究提供了坚实的基础,单细胞转录组数据与公共大队列数据的多组学整合已然成为癌症治疗研究的新热点与此同时,一个关键问题诞生了---如何更好地将两种类型数据进行整合从而挖掘重要癌症的发生发展机制呢?接下来,将给大家介绍两种不同的单细胞转录组数据与大队列bulk转录组数据整合的算法:。
2.转录组测序和单细胞测序区别
BayesPrism和Scissor,同时单细胞相关生信分析需求可以咨询生信人团队生信分析意向表 (mikecrm.com)单细胞转录组测序联合bulk转录组测序(整合篇)一.BayesPrism算法介绍
3.转录组测序和单细胞转录组测序的区别
下面我就先来具体介绍BayesPrism这项整合方法为什么选择这个方法呢?因为我觉得这个方法很有潜力,作者通过比较当前主流的方法如CIBERSORT,证明BayesPrism方法从bulk RNA-seq数据集推断单细胞细胞类型以及对整体基因表达变化的解析上具有比较明显的优势。
4.单个细胞的转录组测序
简单来说,BayesPrism算法以scRNA-seq作为先验信息,从bulk RNA-seq数据集中预测细胞类型的组成和基因表达该文章主要对胶质母细胞瘤(GBM)、头颈部鳞状细胞癌(HNSCC)和皮肤黑色素瘤(SKCM)进行了整合分析,以将细胞类型组成与不同肿瘤类型的临床结果相关联,并探索恶性肿瘤细胞和非恶性细胞状态的空间异质性。
5.单细胞联合转录组找关键基因
本篇文章发表在期刊:Nature Cancer该期刊在最近一年的影响因子: 23.0131. BayesPrism算法研发背景大量研究表明,肿瘤微环境(TME)中恶性细胞和不同类型的非恶性细胞之间的相互作用可以促进血管生成、癌症转移和免疫抑制等。
6.单细胞转录组学技术
非恶性细胞在不同的患者和肿瘤类型之间存在显著差异,某些非恶性细胞群常被用作临床生物标志物和治疗靶点单细胞转录组测序(scRNA-seq)技术的兴起,使得在TME内对单个细胞的转录组进行直接的全基因组测量和表征其异质性成为可能。
7.单细胞基因组与转录组
然而,scRNA-seq的成本和对高质量样本的要求限制了可检测的样本的数量此外,scRNA-seq在细胞捕获中容易受到技术偏差的影响,这妨碍了细胞类型组成的复原为了尽量规避以上问题,研究人员拟从bulk RNA-seq数据中推断单细胞类型组成与基因表达。
8.单细胞转录组测序的原理
然而,现有的反卷积方法没有完全支持异质肿瘤细胞群体中基因表达的预测因此,现有的方法未能解决这些关键问题:在TME中,恶性细胞如何影响非恶性细胞的组成?哪些基因与这些相互作用相关?为了回答这些问题,研究者创建模型,它可以准确地表示每个bulk RNA-seq样本中的细胞类型比例和细胞类型特异性基因表达谱。
9.单细胞转录组测序技术原理
因此,该研究提出了BayesPrism,一个贝叶斯模型,使用scRNA-seq参考表达谱作为先验信息,从bulk RNA-seq数据中推断细胞类型组成和基因表达2. BayesPrism算法推断细胞类型组成和细胞类型特异性基因表达
10.单细胞转录组测序应用
BayesPrism算法具体步骤:BayesPrism使用细胞状态(cell state)、细胞类型、基因列表、bulk RNA-seq数据集样本个数、bulk RNA-seq 表达矩阵以及单细胞表达矩阵作为输入文件,先模拟出每个细胞状态的基因表达矩阵和每个细胞状态的比例,最后通过对每种细胞状态的求和,估算出细胞类型的比例和各细胞类型特异的基因表达水平。
为了验证BayesPrism估计细胞类型比例的效能,作者生成模拟数据,并发现BayesPrism在估计恶性肿瘤细胞比例方面比CIBERSORTx的效果更好(图1 c, d)同时,以PBMC scRNA-seq数据为参考数据集,相对于经典的方法如CIBERSORTx S mode和MuSiC, BayesPrism 对B细胞、髓系细胞、CD4+ T 细胞、CD8+ T细胞以及NK细胞进行了更准确的细胞类型估计(图1e,f)。
总之,这些数据分析表明BayesPrism完善了现在的反卷积算法的性能该研究在恶性细胞中使用BayesPrism算法,进行跨患者异质性的基因表达的预测首先使用了来自8个GBM的scRNA-seq作为参考数据,估计了GBM bulk RNA-seq数据中的细胞类型和基因表达,并发现bulk RNA-seq数据中恶性细胞的基因表达的估计与已知的真实表达高度相似(图1g)。
并且研究发现,当肿瘤纯度大于50%,BayesPrism基因表达估计值与已知基因表达真实值的相关性要大于0.95(图1h)这表明BayesPrism评估的基因表达可以准确地从bulk样本中复原恶性细胞中的基因表达,并且,使用BayesPrism对基因表达的估计要比使用CIBERSORTx或无反卷积更准确(图1h)。
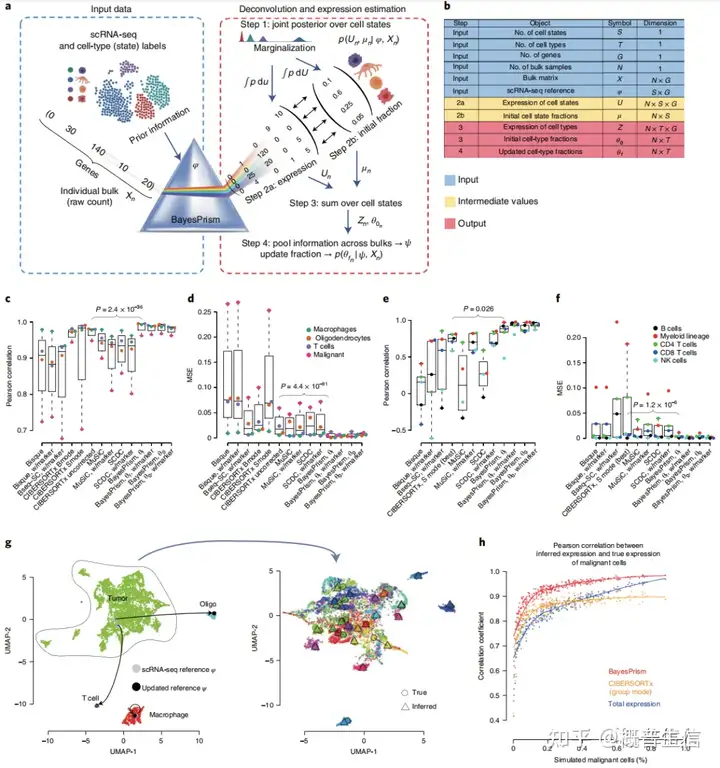
图1:BayesPrism算法流程及性能检验3. 浸润免疫细胞类型和状态对生存的影响该工作分析了TCGA中GBM、HNSCC和SKCM三种肿瘤的1142个bulk样本中细胞类型的比例利用GBM、HNSCC和SKCM三种肿瘤的单细胞参考数据集,估计了6种GBM细胞类型,10种HNSCC细胞类型,8种SKCM细胞类型(图2a)。
接下来,使用两个Cox比例风险模型检查了细胞类型比例和生存率之间的关联,与以往报道一致,在SKCM中,发现CD8+ T细胞和巨噬细胞比例与生存有更强的相关性(图2b,c)并且,根据M1和M2两个巨噬细胞亚群的标记基因, 发现而来自SKCM的M2型巨噬细胞评分最低,M1型巨噬细胞评分与来自HNSCC的巨噬细胞无显著差异(图2d)。
此外,在SKCM中,巨噬细胞高M1极化低M2极化状态与生存率有极强的相关性(图2e)这些发现表明了巨噬细胞比例以及巨噬细胞状态对不同恶性肿瘤具有不同临床结果的影响
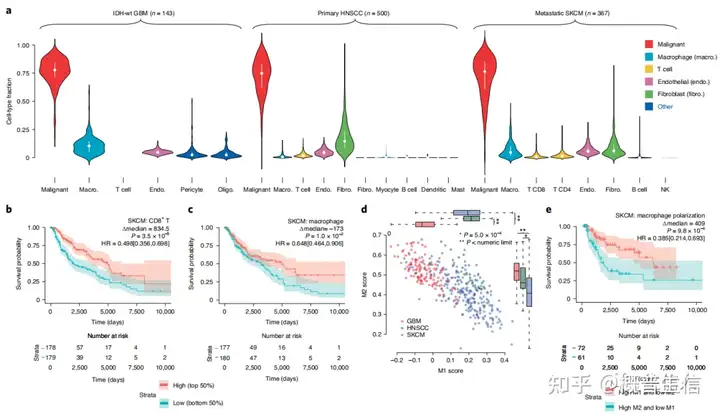
图2:GBM、HNSCC和SKCM的预后与非恶性细胞的细胞类型比例或细胞状态的关系4. BayesPrism识别恶性细胞固有基因程序作者在BayesPrism中开发了一个模块,在剔除非恶性细胞类型的基因表达后,用来推断恶性细胞固有基因程序,该基因程序可以更好地解释bulk RNA-seq数据中的表达异质性(图3a)。
BayesPrism模拟得到的基因程序与对恶性细胞进行因子分解得到的基因程序基本一致(图3b)BayesPrism识别到的每个基因程序的权重与每个肿瘤中分配给每个主要亚型的细胞比例相关(图3c,d)BayesPrism发现GBM中的几个程序与以往的研究相似,包括program 3(classical和AC-like亚型),program 4(mesenchymal)和progam 5(proneural, OPC和NPC-like)(图3e)。
在HNSCC中,program 1富集了以往单细胞研究确定的Partial EMT亚型(图3f)在SKCM中,发现了多个生存相关的基因程序,以及一个T细胞排斥程序(图3g-j)

图3:BayesPrism重新定义GBM分子亚型5. GBM基因程序和细胞类型的空间异质性作者将122个样本的bulk RNA-seq GBM数据集解析为成5个空间结构: LE、IT、CT、MVP和PAN(图4a)。
值得注意的是,已知这些不同结构的TME在血液供应、氧水平和免疫应激这几个方面也呈现很大的不同,所有这些改变都可能影响细胞类型组成和恶性细胞状态利用GBM scRNA-seq数据进行了反卷积,检查在解剖结构中,哪些细胞类型和基因程序富集(图4b,c)。
根据相应的解剖结构,正如预期的那样,MVP区域在内皮细胞和周细胞中高度富集,而LE和IT区域在少突胶质细胞和神经元中富集值得注意的是,PAN区域在巨噬细胞和T细胞中富集将解剖结构与基因程序联系起来,发现LE和IT区域在program 1和2中富集,CT区域在program 3中富集,PAN区域在program 4和program 5中富集,MVP区域在program 4和program 5中富集。
并且发现,CT和MVP区域具有高度的增殖能力,与它们在program 3和program 5中的富集一致,这些program在细胞增殖中富集MVP和PAN都富含组织重塑和免疫相互作用(program 4),而MVP更具有血管生成性,PAN更具有炎症性。
IT和LE都是富集了呼吸链复合物组装通路的,LE是具有最强的呼吸链复合物组装能力、但是具有较低的增殖能力,这解释了它们在program 1中的富集IT也在促炎免疫过程中富集,尤其是干扰素反应(图4 d)。
综上所述,BayesPrism可以将基因程序与空间解剖结构联系起来

图4:BayesPrism揭示了GBM的空间异质性6.小结在这里,作者通过开发一个严格的统计模型来整合scRNA-seq和bulk RNA-seq数据,进行综合分析也为疾病进展提供了新的见解以GBM为例,联合分析bulk RNA-seq队列和空间解剖数据,提出了一个将恶性细胞状态和非恶性细胞浸润与肿瘤进展联系起来的模型(图4e)。
当恶性细胞快速生长时,它们会消耗营养物质,也可能遇到免疫压力,导致坏死(图4e)与此相一致,观察到免疫细胞和间充质program 4的富集,在PAN区域显示更强的间充质激活和更低的呼吸活性(图4b,c)。
恶性细胞可能激活这些组织重塑通路,促进M2巨噬细胞极化和血管生成随着微血管结构的发展,恶性细胞迅速增殖(图4e),MVP附近恶性细胞的细胞周期评分较高(图4d)增殖细胞侵入邻近的正常脑组织,那里的氧气供应充足。
在此过程中,它们的主要任务从快速增殖转变为呼吸作用,以产生合成必要分子机制所需的ATP(图4e)这一发现是基于LE和IT结构中呼吸通路的富集(图4d)最后,随着局部氧水平的降低,恶性细胞恢复快速增殖该研究还表明,program 3可能反映了血液供应充足的癌症生长的早期阶段。
综上所述,该模型说明了GBM细胞如何重塑和响应局部微环境的变化以适应自身生长与以前的方法相比,BayesPrism更准确地将bulk RNA-seq解析为细胞类型的比例和基因表达,从而深入了解肿瘤与微环境的相互作用。
二.Scissor算法介绍下面我再具体介绍一下Scissor这项整合方法为什么选择这个方法呢?因为我觉得这个方法很具有创新性,作者开发了Scissor算法根据bulk RNA-seq数据的表型信息从单细胞数据识别特定的细胞亚群。
在肺癌scRNA-seq数据集中,Scissor确定了与生存率较差和TP53突变相关的细胞亚群在黑素瘤中,Scissor发现一个与免疫治疗应答相关的低PDCD1/CTLA4和高TCF7表达的T细胞亚群本篇文章发表在期刊: Nature Biotechnology 该期刊在最近一年的影响因子: 54.908,水平很高。
1. Scissor算法研发背景单细胞测序技术使复杂组织细胞的综合表征成为可能,从而使生物医学研究和临床实践发生了革命性的变化与测量整个组织平均特性的bulk数据相比,scRNA-seq允许在异质组织生态系统中识别不同细胞亚群的细胞类型和状态。
要从单细胞数据中识别关键亚群,标准方法是执行无监督聚类来定义细胞群,并评估已知细胞类型和通路中标记基因的富集情况,以评估每个细胞集群的重要性然而,识别驱动表型(如疾病阶段、肿瘤转移、治疗反应和生存结果)的细胞亚群具有不可缺少的重要性,因为它将促进细胞类型靶向治疗和预后生物标志物的发现。
Scissor的新颖之处在于,它使用来自bulk数据的表型信息来识别与疾病高度相关的细胞亚群,进而揭示疾病机制,提高疾病的诊断和治疗2. Scissor算法基本流程Scissor算法具体步骤:Scissor使用单细胞表达矩阵、bulk表达矩阵和感兴趣的表型(图5a)。
每个样本的表型注释可以是一个连续的因变量、二元分类向量或临床生存数据Scissor的关键步骤是量化单细胞数据和批量数据之间的相似性,通过测量,如每对细胞和批量样本的皮尔逊相关性在这之后,剪刀优化与样本表型相关矩阵的回归模型(图5b)。
回归模型的选择取决于输入表型的类型,例如,连续变量的线性回归,二分变量的logistic回归和临床生存数据的Cox回归根据估计回归系数的符号,系数为非零的细胞可表示为剪刀阳性(Scissor+)细胞和剪刀阴性(Scissor−)细胞,它们分别与感兴趣的表型呈正相关和负相关(图5c)。
此外,为了控制单细胞和bulk数据之间的虚假关联,设计了一个可靠性显著性检验,以确定所选数据是否适合的表型-细胞关联(图5d)最后,Scissor选择的细胞将在下游分析中进一步表征,如特征基因和功能富集途径的探索(图5e)。
首先评估了Scissor在一系列模拟数据集上的性能,结果表明Scissor识别的与已知的表型相关的细胞亚群与真实结果在很大程度上是一致的(图5f, g, h)3.识别正常和肿瘤表型相关的细胞亚群使用577个TCGA-LUAD bulk样本中的肿瘤和正常表型对肺癌单细胞数据进行Scissor分析。
在29,888个来自不同细胞类型的细胞中(图5i), Scissor选择了361个Scissor+细胞和534个Scissor−细胞,它们与肿瘤和正常表型具有高置信关系(图5j)正如预期,超过98%的Scissor+细胞被证实为恶性细胞(图5k)。
至于Scissor−细胞,主要分布于非恶性细胞类型中(图5k)髓系细胞和肺泡细胞是两种主要选择的细胞类型,分别占Scissor−细胞总数的42.3%和36.9%剪刀细胞中的所有细胞类型,尤其是肺泡细胞,都是正常肺组织中的重要细胞类型。
因此,这些分析证明了Scissor可以从单细胞数据中准确地识别出表型相关的细胞
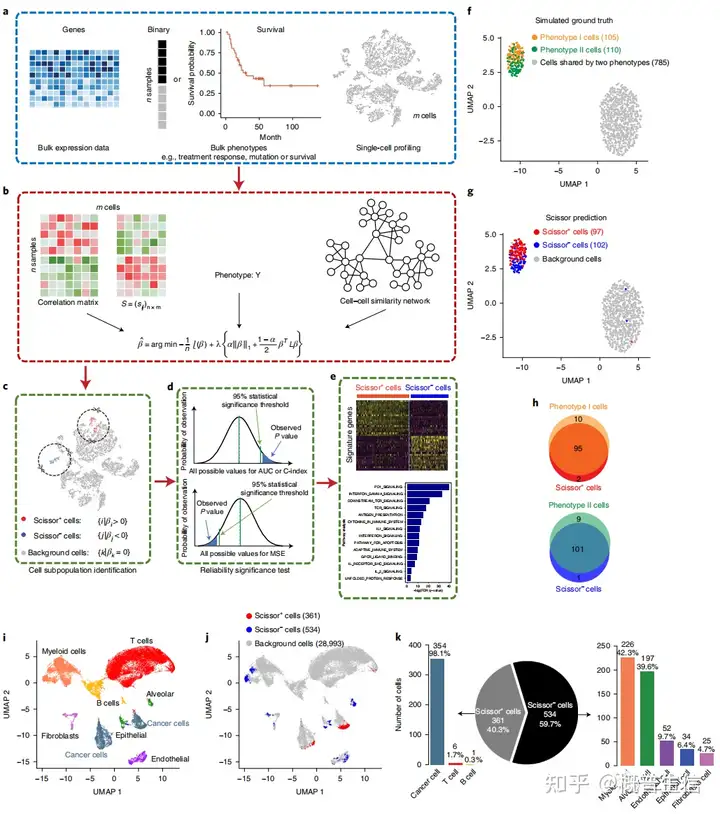
图5:Scissor的工作流程以及应用效能4. 发现低氧亚群与较差的生存使用Scissor,基于带有生存信息的471个TCGA-LUAD bulk样本为识别肺癌scRNA-seq数据集中具有侵袭性的癌细胞亚群。
这些细胞被分离成12个Cluster(图6a)在205个Scissor选择的细胞中,201个Scissor+细胞与较差的存活率相关(定义为Scissor_WS细胞),只有4个Scissor−细胞与良好的存活率相关(图6b)。
Scissor_WS细胞为主要来自Cluster 1和Cluster 3(图6c)差异基因分析显示,与其他所有细胞相比,Scissor_WS细胞中分别有23个上调基因(有多个重要的缺氧相关基因)和205个下调基因差异表达(图6d,e)。
功能富集分析也证实了缺氧相关的通路,如糖酵解和糖代谢通路在Scissor_WS细胞中被激活(图6f)并发现在独立的GEO数据集中,特征分数高的患者预后明显差于签名分数低的患者(图6g)并在单因素Cox生存分析中,发现只有病理分期和Scissor_WS特征与患者生存显著相关(图6h)。
此外,在对多变量Cox生存分析中的肿瘤分期进行调整后,Scissor_WS特征在两个数据集中仍然具有统计学意义(图6i)总之,Scissor算法从LUAD scRNA-seq数据中发现了一个具有侵袭性的癌细胞亚群,该亚群与较差的生存结果相关,其特征是缺氧相关基因的过度表达。
高度缺氧信号可能会推动LUAD进展,从而给肿瘤中含有大量此类细胞的患者带来不良结果
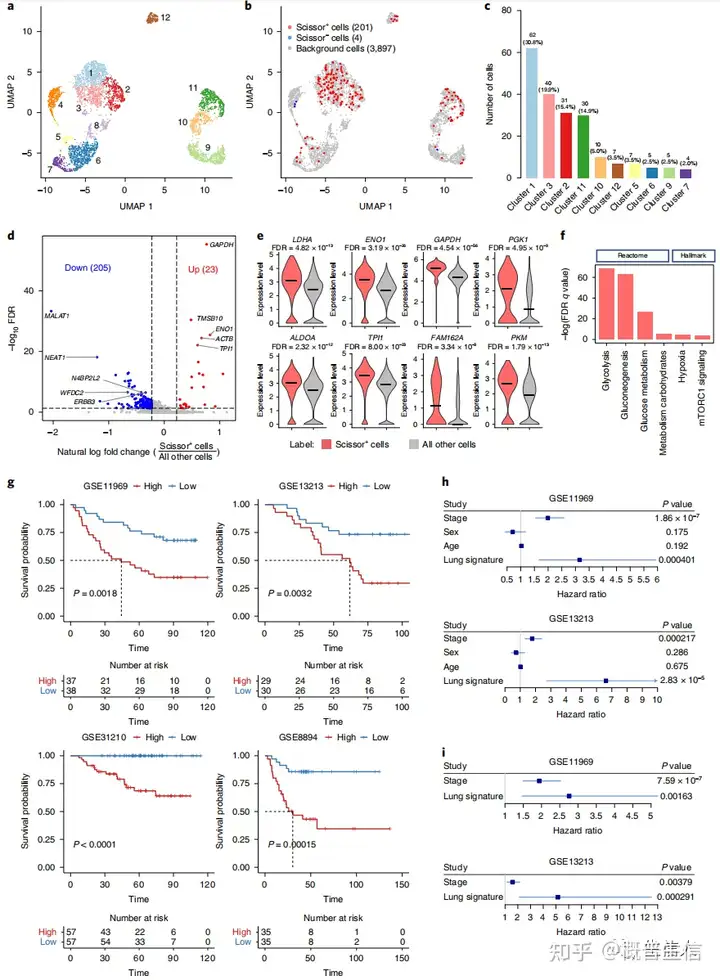
图6:以TCGA-LUAD生存结果为指导的肺癌细胞Scissor鉴定结果5. 分析与TP53突变相关的细胞亚群基于TCGA-LUADTP53突变状态(突变型或野生型)作为表型特征来指导对肺癌单细胞数据的细胞亚群的鉴定。
Scissor共鉴定了414个与TP53突变相关的Scissor+细胞和318个与野生型相关的Scissor−细胞(图7a)差异基因分析显示Scissor+细胞上调337个基因包括E2F靶基因和细胞周期进展相关基因,如AURKA、CDK1、CCNB2和TOP2A(图7b)。
功能富集分析也证实了E2F等细胞周期相关的通路在Scissor+细胞中被激活(图7c)转录因子分析显示,在Scissor+细胞中,E2F转录因子家族成员E2F1和E2F4的活性均显著升高(图7d),然而TP53在Scissor+细胞中是失活的(图7d)。
此外,通过关联这337个上调基因(定义为TP53突变特征)与临床结果,证明TP53突变评分较高的患者预后明显较差(图7e)该研究还发现MHC类相关基因HLA-A、B2M和CD74在Scissor+细胞中均下调(图7f)。
并已有报道称在免疫治疗耐药的癌症患者中B2M的功能丧失突变因此,Scissor的表型分析表明TP53突变可能是检查点抑制剂治疗耐药性的一个机制
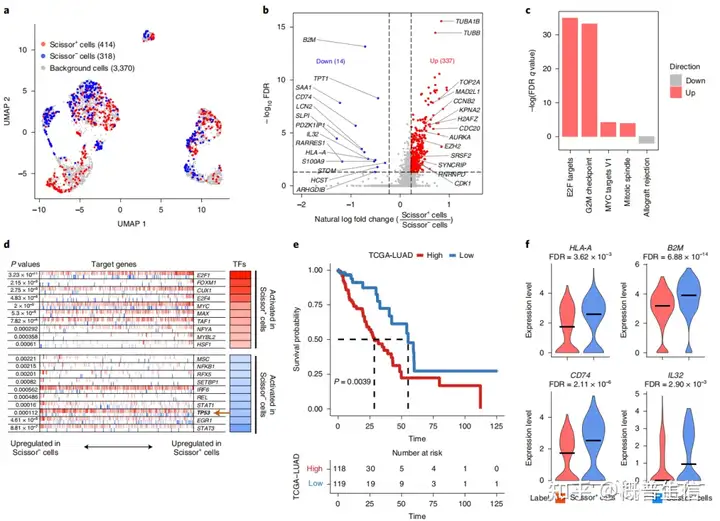
图7:基于TP53突变状态的肺癌细胞Scissor鉴定结果6.鉴定与免疫治疗相关的T细胞亚群为了了解免疫检查点阻断治疗(ICB)反应的机制,对黑素瘤scRNA-seq数据集中的T细胞以及已知免疫治疗反应信息的黑素瘤患者的bulk RNA-seq数据进行了Scissor分析,以识别与ICB反应相关的T细胞亚群。
在scRNA-seq数据分析中,这些T细胞被聚集成6个Cluster(图8a)通过使用Scissor算法,确定了105个T细胞为Scissor+细胞,这些细胞与良好的免疫治疗反应相关(定义为Scissor_FR细胞)(图8b),这105个Scissor_FR细胞主要分布在Cluster 2和Cluster 3中(图8c)。
差异基因分析表明Scissor_FR细胞增加了与T细胞记忆相关基因(CCR7, SELL和IL7R)的表达以及低表达抑制基因(HAVCR2, LAG3, PDCD1和CTLA4)和MHC II类基因(HLA-DRB5, HLA-DRB1, HLA-DPA1, HLA-DQB2和HLA-DRB6)(图8d,e)。
同时,Scissor_FR细胞也表现出转录因子TCF7的表达增强,这与ICB治疗的良好结果相关(图8e)此外,富集分析显示,Scissor_FR细胞TNF-α信号通路较高,CTLA4、PD1信号通路活性较低(图8f)。
以上与有效免疫治疗应答相关的差异表达基因(定义为免疫治疗应答特征)可以用于预测治疗应答率该研究发现有ICB应答者的签名分数明显高于无ICB应答者(图8g)此外,免疫治疗应答信号中上调和下调的基因在应答者和非应答者中也显著富集(图8h)。
进一步评估了五种不同分化状态的肿瘤浸润淋巴细胞的免疫治疗反应特征,发现LAG3-low/PD1-low效应CD8 T细胞和记忆前体CD8 T细胞的特征评分最高(图8i,j)这一结果表明,Scissor_FR细胞更像PD1-low记忆前体细胞,具有较高的TCF7表达,并与良好的免疫治疗反应有关。
总之,对黑色素瘤scRNA-seq数据集的Scissor分析独立揭示了PDCD1/CTLA4低和TCF7高T细胞亚群,其独特的转录组特征对免疫治疗的良好反应至关重要
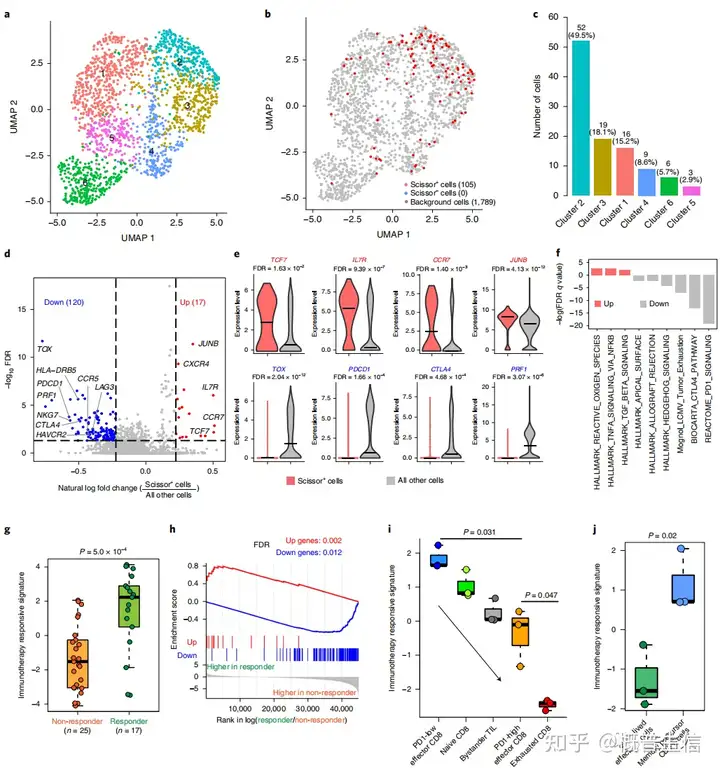
图8:黑色素瘤ICB治疗应答相关T细胞的Scissor识别结果三.小编总结通过使用BayesPrism算法整合,既可以解析大样本队列的细胞类型的比例,又能获得细胞特异的基因表达,通过使用Scissor算法整合,可以获得与临床表型紧密相关的细胞群体。
总而言之,单细胞转录组数据和bulk转录组数据:优势互补,将单细胞转录组数据和bulk转录组数据进行有效结合,将以全新研究思路出发,会发现更多未知且精细化结果单细胞相关生信分析需求可填写生信意向表生信分析更多精彩好文
生信分析热点分享:7+免疫治疗切入点 - 知乎 (zhihu.com)SCI生信分析: 调节性T细胞对免疫相关不良事件(irAEs)的重要性 - 知乎 (zhihu.com)生信分析 ▎单细胞分型方法怎么选? - 知乎 (zhihu.com)
以上就是关于《算法:单细胞转录组测序联合bulk转录组测序_单核转录组测序》的全部内容,本文网址:https://www.7ca.cn/baike/13611.shtml,如对您有帮助可以分享给好友,谢谢。